Современное лечение гемофилии
| ГЕМОФИЛИЯ Гемофилия — наиболее часто встречающееся наследственное патологическое состояние, связанное с нарушением I фазы свертывания крови — образования плазменного тромбопластина. Классификация. Различают несколько типов гемофилии: 1) гемофилия А (классическая), обусловлена дефицитом фактора VIII (антигемофильный глобулин); ген, кодирующий синтез фактора VIII, находится на Х-хромосоме; мутация этого гена (возникновение гена гемофилии А) приводит к резкому нарушению синтеза фактора VIII с развитием его дефицита; 2) гемофилия В (болезнь Кристмаса) — наследственный дефицит IX фактора свертывания крови (фактора Кристмаса); частота этого заболевания среди других вариантов гемофилии составляет 6—20%; 3) гемофилия С — наследуемый аутосомно дефицит фактора XI; редкий вариант гемофилии; его частота встречаемости составляет до 3%; 4) гемофилия А + В — очень редкий вариант (частота встречаемости до 1,5%) сочетанного дефицита факторов VIII и IX. В настоящем разделе будет подробно рассмотрен наиболее часто встречающийся вариант гемофилии — гемофилия А. Эпидемиология. В среднем 10 случаев на 100 000 жителей мужского пола. Этиология. Гемофилия А и В — рецессивно наследуемые, сцепленные с полом (Х-хромосомой) заболевания; болеют лица мужского пола, женщины передают заболевание. Генетические дефекты характеризуются недостаточным синтезом или аномалией факторов VIII (коагуляционная часть) — гемофилия А, или фактора IX — гемофилия В. Временный (от нескольких недель до нескольких месяцев) приобретенный дефицит факторов VIII, реже — IX, сопровождающийся сильной кровоточивостью, наблюдается и у мужчин, и у женщин (особенно в послеродовом периоде, у лиц с иммунными заболеваниями) вследствие появления в крови в высоком титре антител к этим факторам. Локализующийся в Х-хромосоме ген гемофилии передается от больного гемофилией всем его дочерям, и они в дальнейшем передают этот ген своим потомкам. Все сыновья больного остаются здоровыми, так как получают единственную Х-хромосому от здоровой матери. Женщины – кондукторы заболевания, имеющие вторую нормальную Х-хромосому, как правило, не страдают кровоточивостью, но активность фактора VIII у них снижена в среднем в 2 раза. У половины (50%) сыновей этих женщин, имеющих ген гемофилии, есть шансы родиться больными (при равной возможности получить от матери патологическую или нормальную Х-хромосому), а половина (50%) дочерей – стать передатчицами болезни. 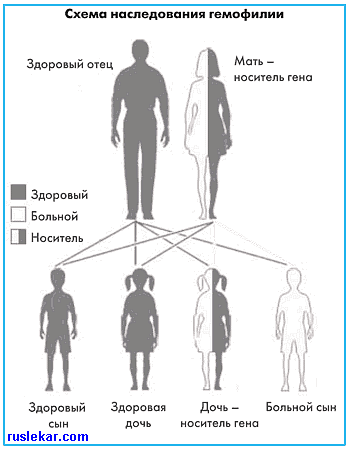 Патогенез. В основе кровоточивости лежит изолированное нарушение начального этапа внутреннего механизма свертывания крови, вследствие чего резко удлиняется общее время свертывания цельной крови (в том числе параметр R тромбоэластограммы), и меняются показатели более чувствительных тестов — аутокоагуляционного (АКТ), активированного парциального тромбопластинового времени (АПТВ) и т. д. Протромбиновое время (индекс) и конечный этап свертывания, а также все параметры тромбоцитарного гемостаза (число тромбоцитов и все виды их агрегации) не нарушаются. Пробы на ломкость микрососудов (манжеточная и др.) остаются нормальными. Степень тяжести гемофилии определяется по уровню активности фактора VIII (IX) в крови в процентах от нормального (норма для фактора VIII – 56-110%): • тяжелая форма – < 2,0%; • гемофилия средней тяжести – уровень фактора 2,1-5,0%; • легкая форма – > 5,0%. По классификации З. Баркагана (1980), широко используемой ранее, дополнительно выделяются «скрытая» и «крайне тяжелая» формы с уровнями активности фактора 13-55% и 0-1% соответственно. Клиническая картина представлена геморрагическим диатезом гематомного типа уже в детском возрасте. При повреждении тканей имеют место длительные кровотечения, формируются гематомы, гемартрозы. Тяжесть геморрагических проявлений при гемофилии А строго коррелирует с дефицитом VIII фактора в плазме крови, уровень которого зависит от функционального состояния гена, кодирующего этот фактор, и является, таким образом, генетически запрограммированным. Диагностика. Легкая форма гемофилии может быть впервые распознана в зрелом возрасте во время оперативного вмешательства. Тяжелую или умеренную гемофилию предполагают у мальчиков с гематомным типом кровоточивости и гемартрозами, возникающими с раннего детства. Лабораторные признаки: удлинение АЧТВ при нормальных показателях времени кровотечения, количества тромбоцитов, протромбиновое время, ПК и уменьшения факторов VIII или IX. В случаях с низким содержанием VIII фактора необходимо исключить болезнь Виллебранда. Для дифференциальной диагностики гемофилии А и В используют тест генерации тромбопластина, коррекционные пробы в аутокоагулограмме — при гемофилии А нарушение свертываемости устраняется добавлением к плазме больного донорской плазмы, предварительно адсорбированной сульфатом бария (при этом удаляется фактор IX, но сохраняется фактор VIII), но не устраняется нормальной сывороткой, продолжительность хранения которой 1—2 сут (содержит фактор IX, но лишена фактора VIII); при гемофилии В коррекцию дает старая сыворотка, но не BaS04-плазма. При наличии в крови больного иммунного ингибитора антигемофилического фактора («ингибиторная» форма гемофилии) коррекцию не дают ни BaS04-плазa, ни старая сыворотка, мало нарастает уровень дефицитного фактора в плазме больного после в/в введения его концентрата или донорской плазмы. Титр ингибитора определяют по способности разных разведений плазмы больного нарушать свертываемость свежей нормальной донорской плазмы. Кофакторная (компонентная) гемофилия — очень редкая форма. Тип наследования аутосомный. Низкая активность фактора VIII устраняется в тесте смешивания плазмы исследуемого с плазмой больного гемофилией А. Время кровотечения, адгезивность тромбоцитов, уровень в плазме фактора Виллебранда и его мультимерная структура не нарушены, что отличает кофакторную гемофилию от болезни Виллебранда. Пренатальная диагностика. 1. Исследование ворсин хориона (10 нед беременности) в случае известной мутации требует исследования матери и по крайней мере одного больного родственника. 2. Исследование фетальной крови (18—20 нед беременности) связано с риском гибели плода (2-5%). Дифференциальную диагностику гемофилии и других вариантов коагулопатий (дефицит V, VII, X или XI факторов, гипофибриногенемии, болезни Виллебранда) проводят на основании коррекционных проб, выявляющих дефицит конкретного фактора плазменного звена гемостаза у больного. Дифференциальный диагноз с тромбоастениями (Гланимана) основывается на исследовании функции тромбоцитов и определении содержания факторов свертывания. Лечение и профилактика геморрагических проявлений при гемофилии А и В. Заместительная терапия. Используют концентраты VIII или IX фактора, полученные из плазмы человека (криопреципитат антигемофильного глобулина; антигемофильная плазма; свежезаготовленная плазма до 4 ч хранения; свежезаготовленная кровь до 24 ч хранения) или рекомбинантные. Эффективно прямое переливание крови или переливание свежеприготовленной крови. Однако эти варианты лечения и применение препаратов, получаемых из донорской крови, сопряжены с риском заражения вирусами гепатита В и С, цитомегаловирусом, ВИЧ (необходима вирусная инактивация). Многочисленные повторные трансфузии могут привести к сенсибилизации больного и вызвать у него выработку AT против вводимого VIII фактора. Поэтому лучше использовать рекомбинантный препарат VIII фактора, который на сегодняшний день является оптимальной лекарственной формой для коррекции гемофилии А, и рекомбинантный IX фактор при гемофилии В. Доза препаратов зависит от исходного уровня этого фактора в плазме больного, а также от стоящей перед врачом задачи. Для обеспечения надежного гемостаза у больного с одним гемартрозом, одной гематомой, дешевым кровотечением, кровотечением из лунки одного удаленного зуба или кровотечением из небольшой раны необходимо добиться повышения концентрации VIII фактора до 10—20% от нормального уровня. У больного с несколькими гемартрозами, большими гематомами, заглоточной гематомой, гематомой в области шеи, гематораксом, желудочно-кишечным кровотечением, гематурией, кровоизлиянием в ЦНС, с кровотечением из больших ран или с переломами необходимо добиться повышения концентрации VIII фактора до 20-40% от нормального. Больным с гемофилией А, которым предстоит оперативное вмешательство, концентрацию VIII фактора поднимают до 50-70% от нормального уровня. Десмопрессин стимулирует эндогенное высвобождение VIII фактора и в некоторых случаях используется при гемофилии А. Кровоизлияние в мягкие ткани может осложняться нагноением гематомы. В этих случаях показана антибактериальная терапия антибиотиками широкого спектра действия. При этом следует учитывать, что при гемофилии любые в/м инъекции противопоказаны, так как они могут стать причиной образования обширных гематом. Все необходимые инъекции выполняют в/в. Местная терапия. При купировании наружных кровотечений в дополнение к обязательной заместительной терапии препаратами VIII фактора можно использовать местные воздействия — обработать место кровоточивости тромбином, тромбопластином, е-аминокапроновой кислотой, используют транексамоновую кислоту. При необходимости на рану накладывают швы и давящую повязку. Лечение гемартрозов. В остром периоде — возможна более ранняя заместительная гемостатическая терапия в течение 5-10 дней, при больших кровоизлияниях — пункция сустава с аспирацией крови и введением в его полость гидрокортизона или преднизолона (при строгом соблюдении асептики). Иммобилизация пораженной конечности на 3—4 дня, затем — ранняя лечебная физкультура под прикрытием криопреципитата; физиотерапевтическое лечение, в холодном периоде — грязелечение (в первые дни под прикрытием криопреципитата). При всех кровотечениях, кроме почечных, показан прием внутрь е-аминокапроновой кислоты по 4—12 г/сут (в 6 приемов). Локальная гемостатическая терапия: аппликация на кровоточащую поверхность тромбина с гемостатической губкой и е-аминокапроновой кислотой. Викасол и препараты кальция неэффективны и не показаны. Остеоартрозы, контрактуры, патологические переломы, псевдоопухоли требуют восстановительного хирургического и ортопедического лечения в специализированных отделениях. При артралгиях противопоказано назначение нестероидных противовоспалительных препаратов, резко усиливающих кровоточивость. На фоне терапии концентратами VIII фактора у 30% больных гемофилией А появляется устойчивость к терапии вследствие выработки антител (ингибиторов), блокирующих прокоагулянтную активность вводимых факторов. При гемофилии В частота развития ингибиторов меньше. Способы преодоления ингибиторной активности: — повышенные дозы фактора; — препараты шунтирующего действия (свиной фактор VIII, иммуноабсорбция с последующим назначением VIII или IX фактора, рекомбинантный концентрат активированного VII фактора и активированный концентрат протромбинового комплекса (АРСС), иммуномодуляторы (низкие дозы циклофосфамида, в/в у-глобулин). Прогноз. При адекватной заместительной терапии препаратом VIII фактора (особенно рекомбинантным фактором VIII) прогноз у больных с гемофилией А достаточно благоприятный. Профилактика. 1. Медико-генетическое консультирование, определение пола плода и наличия в его клетках гемофилической Х-хромосомы. 2. Профилактика гемартрозов и других геморрагии: диспансеризация бальных, рекомендации определенного образа жизни, устраняющего возможность травм, раннее использование допустимых видов лечебной физкультуры (плавание, атравматичные тренажеры). Организация раннего введения антигемофилических препаратов на дому (выездные специализированные бригады, обучение родителей в школе медицинских сестер); рентгенотерапия пораженных суставов, хирургическая и изотопная синовэктомия. В наиболее тяжелых случаях систематическое (2—3 раза в мес) профилактическое введение концентрата факторов VIII или IX. Перспективы Несмотря на серьезность этого заболевания, у пациентов с гемофилией есть основания для оптимизма. Со стороны исследователей и пациентов этой проблеме еще никогда не уделялось такого внимания, как сейчас. Так, в частности: – специалисты Медицинского центра в Бостоне разработали методику электроимпульсного внедрения здорового гена в клетки, выделенные из организма больного: в клетки кожи больных тяжелой формой гемофилии, взятые из области предплечья, с помощью короткого электрического импульса был введен ген фактора свертывания крови VIII. Затем эти генетически модифицированные клетки были введены в подкожно-жировой слой пациентов. В катамнезе у 30% больных в течение 10 месяцев отсутствовали спонтанные кровотечения, ранее отмечавшиеся часто; у 70% пациентов уровень фактора VIII в крови значительно повысился, что позволило снизить дозы препаратов заместительной терапии. Данное исследование – еще один шаг на пути к медицине будущего, а именно к медицине, ориентированной на причину болезни – дефектный ген; – не прекращаются исследования касательно получения рекомбинантных факторов. Так, например, в клинической практике успешно используются факторы свертывания крови VIIа (НовоСевен) и VIII (Коджинейт), получаемые из культуры линии клеток почек новорожденного хомяка. Кроме того, в некоторых экспериментах показана возможность получения трансгенного коровьего молока, обогащенного фактором. | |
|
| |
| Просмотров: 11929 | |
| Всего комментариев: 0 | |